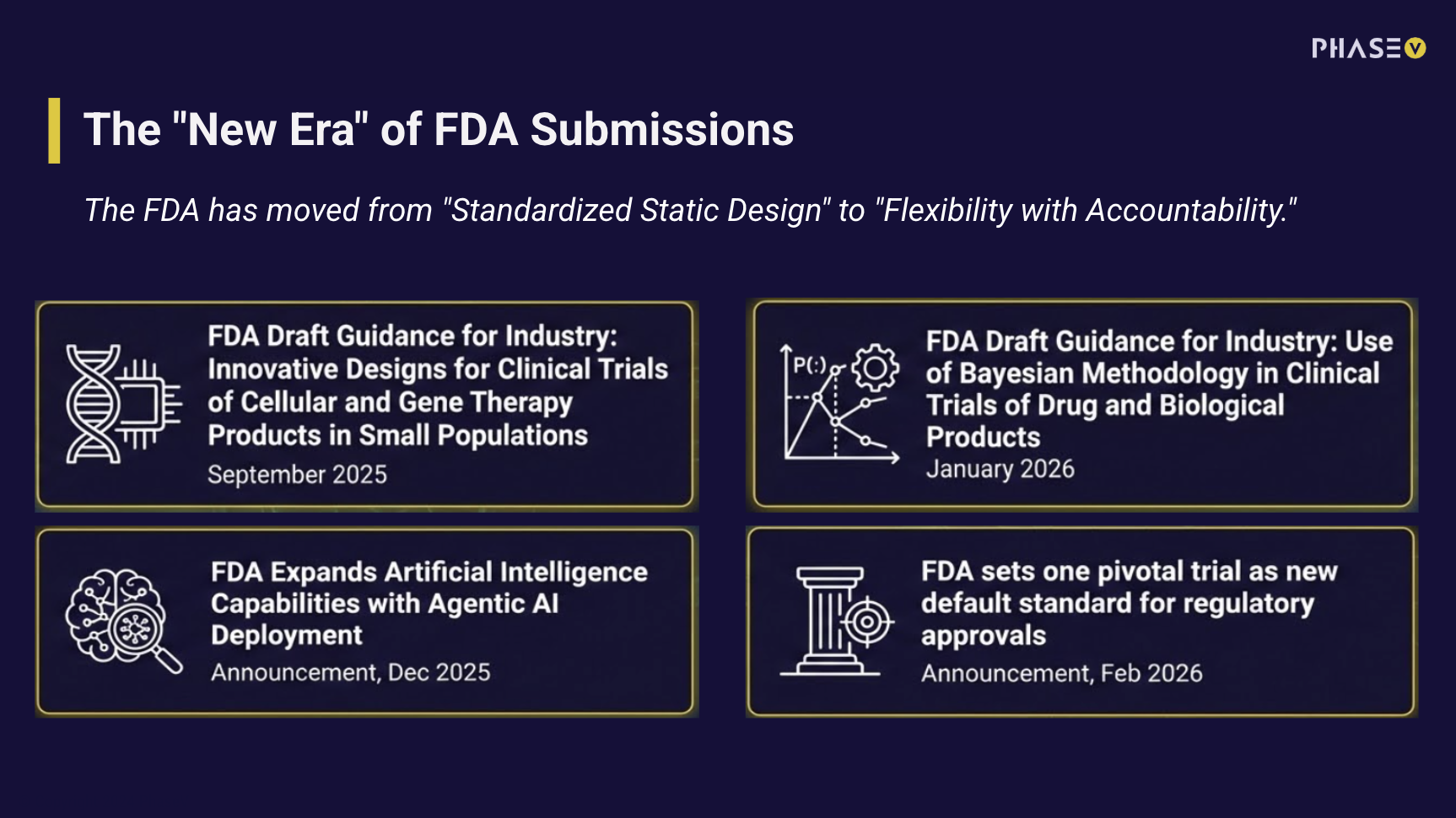
What the latest FDA updates mean for the future of Clinical Trial Design

Recent FDA updates are signaling a meaningful shift in how clinical trials can be designed, analyzed, and approved. A recent PhaseV webinar brought together industry leaders to unpack these changes - from Bayesian methods to AI-enabled review processes - and what they mean for sponsors navigating an increasingly complex regulatory landscape.
1. Bayesian methods are moving toward the mainstream
One of the most notable developments is the FDA’s growing openness to Bayesian approaches. Historically limited to niche use cases, Bayesian methods are now gaining broader regulatory acceptance. The new Bayesian guidance document now even explicitly allows posterior probabilities to replace traditional p-values as the primary evidence standard. This is especially helpful in trials that borrow strength from historical data, for which traditional metrics such as Type I error are less meaningful.
Luis Peña (Founder and CEO of Evommune) highlighted the real-world impact of this shift, describing how Bayesian design enabled stronger decision-making in a Phase 2a trial at his company:
“You tend to reduce the risk and get a more decisive answer with a smaller trial […] we get a more definitive go/no-go decision going forward.”
By incorporating historical control data, his team increased statistical power while only minimally increasing Type I error - ultimately delivering a clear efficacy signal.
Fabio Rigat, PhD (Executive Director, Oncology and GI/GU/GYN Head of Strategy at AstraZeneca) reinforced that Bayesian approaches are already proving valuable, especially in early development:
“Bayesian methods […] incorporate multifactorial, multiple endpoints to robustify decision making.”
However, he also noted that while they are becoming standard in early phase studies, broader adoption of Bayesian methods in registrational trials in non-rare disease is still evolving.
2. External data and “Digital Twins” can improve trial efficiency
Beyond Bayesian borrowing, the panel also explored how real-world and historical data can be operationalized through approaches like digital twins.
Feng Gao (Sr. Director, Head of Innovative Data Analytics, Biogen) explained how prognostic modeling using historical placebo data can significantly improve trial efficiency:
“You can either increase the power or reduce the sample size […] we were able to reduce the sample size by close to 20% without impacting power.”
Rather than fully replacing control arms, these digital twins act as “super covariates,” helping explain patient variability and improving statistical precision. Still, Rigat cautioned against overreliance:
“There is a promise, but there’s also a risk […] in assuming that everything that you’ve seen in the past is enough to accurately predict the future.”
3. Trial design is becoming more flexible - especially in Rare Disease
The FDA is also signaling greater flexibility in trial design, particularly for small or genetically defined populations. Sponsors can increasingly consider single-arm trials, partially randomized studies augmented with external controls, and adaptive designs (say, that add or delete doses, or stop early for success or futility).
Rigat emphasized the complexity of applying these approaches in cutting-edge areas like cell and gene therapy:
“There is the ambition of actually bringing CAR-T beyond its original development context, which was hematological malignancies, into solid tumors... that poses a number of questions about their ability to deliver as a single monotherapy, versus the need to actually augment with a combination strategy.”
This reflects a broader trend: innovation in therapy is forcing parallel innovation in evidence generation.
4. AI will reshape how evidence is submitted - and reviewed
Another major theme was the rise of agentic AI in regulatory workflows. As the FDA begins using AI tools to evaluate submissions, sponsors may need to rethink how they structure their data and documentation.
Gao suggested a practical near-term step:
“We should […] pressure test […] what kind of format of your package is AI friendly.”
Looking ahead, he even envisioned a future where both submissions and initial agency reviews are fully automated:
“We just submit all the data… and [the FDA] can use AI to assemble whatever they want.”
5. A shift toward “One Pivotal Trial” - with strong supporting evidence
Finally, the FDA’s move toward a “one pivotal trial” standard places greater emphasis on the totality of evidence. This raises important questions about what qualifies as “decision-grade” support—from real-world data to model-based approaches.
As Gao noted, success will still depend on rigor and early engagement:
“We always have to be prepared […] to understand what is `sufficient evidence’.”
Bottom Line
Across all these changes, a clear pattern emerges: the FDA is expanding the toolkit for clinical development. Bayesian methods, external data, and AI are no longer fringe concepts—they are becoming central to how evidence is generated and evaluated.
For sponsors, the opportunity is significant - but so is the responsibility to apply these tools rigorously, transparently, and in close collaboration with regulators.